
 |
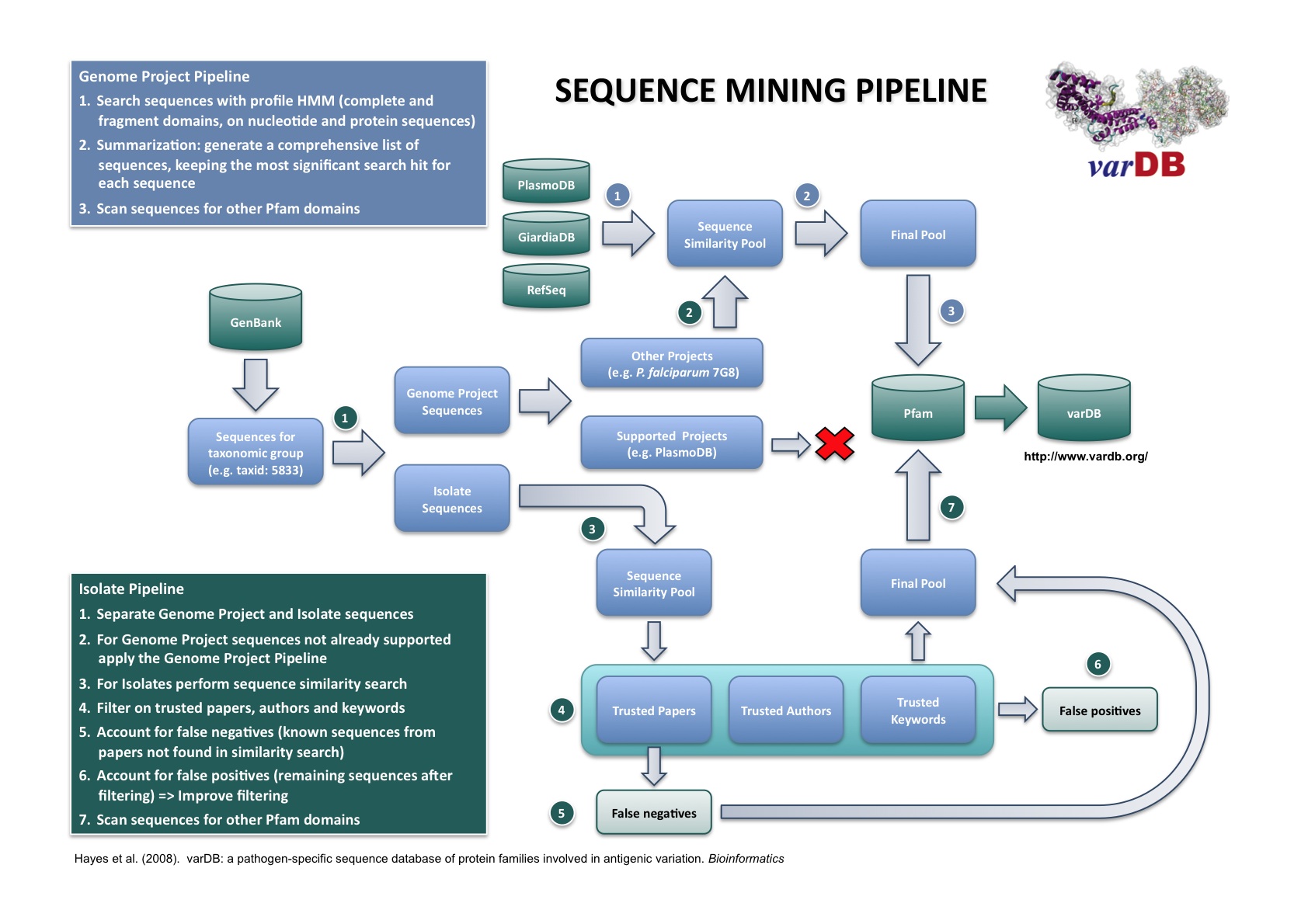
Figure 1. Data mining work flow. |
Pathogens of medical and veterinary importance demonstrating antigenic variation were selected for inclusion in varDB. The identification of antigenic variant gene families was based on published reports in the scientific literature. A hierarchical search strategy encompassing sequence similarity profile, keyword, literature and author specific searches was employed. Sequences and annotations were downloaded from DDBJ/EMBL/GenBank based on taxon-specific identifiers and separated either as isolate-specific or genome project sequences. Isolate sequences were collected based on sequence similarity profiles, and keyword searches based on author, publication and gene/protein annotations. Genome sequences were searched for similarity to the target gene families using HMM models. GenBank records for some genome sequencing projects may be out of date due to progressive re-annotation following initial submission. In this case, current data were retrieved directly from project or species-specific centers (e.g., the Broad Institute Microbial Sequencing Center, GiardiaDB, etc.).
Bacterial and eukaryotic sequences were identified based on matches to Pfam domains belonging to the gene families of interest (Sonnhammer et al, 1997). Genome sequences and other annotation data were downloaded from The Broad Institute (Plasmodium falciparum strains Hb3 and Dd2), PlasmoDB (Plasmodium falciparum 3D7) and NCBI (Anaplasmataceae). Data were transformed to a common format (FASTA format for sequence searches and GFF format for sequence annotations).
Hidden Markov Models (HMM) (Eddy, 1998) were used to identify sequences belonging to the gene families of interest. To ensure that all related sequences were detected (Eddy, 2003), com-plete and fragment models were downloaded from the Pfam data-base (version 22). HMMER (Eddy, 2003) and Genewise (Birney and Durbin, 2000) were use for searching protein and nucleotide sequences, respectively. Sequences were collected based on an E-value cutoff of 1E-02 (Eddy, 2003). A comprehensive list of unique sequences was generated from the four search results, following this priority schedule: hmmer_ls > hmmer_fs > genewise_ls > genewise_fs. The corresponding scores and E-values were kept as annotations included in varDB.
Each selected sequence was then scanned for Pfam domain matches, using the model database Pfam-A (version 22). Both complete and fragment domains were selected based on the same cutoff used for family identification. The position of the domain, the score and the E-value for each hit were stored as annotations. The domain architecture (i.e. the order of the domains in the sequence) was calculated.
When new sequences are added to the database, features are extracted from GenBank flat files as well as from automated feature discovery tools and manual curation based on the literature. Each protein family is aligned using MAFFT (Katoh et al, 2005), followed by manual inspection and adjustment, particularly within hypervariable regions, which are difficult to align automatically. Nucleotide sequences are then codon-aligned based on the protein alignment using RevTrans (Wernersson and Pederson, 2003). Sequence annotations are compiled using sequence analysis tools and annotations from the literature and online databases. Secondary structure predictions are calculated for protein families, and 3D structure and epitope information is incorporated where available.
In the figure above, "Literature" refers to primary research articles that link directly to antigenic variation sequences via accession numbers. "Researchers" are researchers who have previously published papers covering sequences involved in antigenic variation and may be listed as authors on direct submission and unpublished sequence records. Keywords can be shown by manual inspection to reliably and specifically retrieve antigenic variant sequences without retrieving false positives.